Модуль 2 Нуклеофильное замещение у насыщенного атома углерода
Комплексная цель модуля
Систематизировать и углубить знания о механизмах нуклеофильного замещения у насыщенного атома углерода и элиминирования. Получить представление об использовании реакций нуклеофильного замещения у насыщенного атома углерода и элиминирования в органическом синтезе.
Программа семинара
Понятие о реакциях нуклеофильного замещения. Механизмы мономолекулярного и бимолекулярного замещения у насыщенного атома углерода (SN1 и SN2). Факторы, оказывающие влияние на протекание реакций нуклеофильного замещения: строение субстрата, природа реагента, полярность растворителя, катализаторы. Перегруппировки карбокатионов. Реакции элиминирования E2 и Е1.
Краткая теория для подготовки к семинару и решения задач
В реакциях нуклеофильного замещения у насыщенного атома углерода атакующая частица с неподеленной парой электронов (нуклеофил) замещает в молекуле субстрата группу Х, связанную с sp3-гибридизованным атомом углерода. В результате электронная пара, принадлежащая связи С-Х, остается у уходящей группы, а новая связь С-Nu образуется за счет электронной пары нуклеофила:
R-X + Nu: ? R-Nu + X:
Легкость реакций нуклеофильного замещения определяется совокупностью структурных и внешних факторов. В частности, реакции благоприятствует повышенная полярность и поляризуемость связи С-Х в субстрате. Полярность этой связи обеспечивает возникновение на атоме углерода значительного положительного заряда, что облегчает его атаку нуклеофилом. Поляризуемость – это дополнительная полярзация связи С-Х, но уже под действием электрического поля приближающегося реагента, т. е. в момент самой реакции.
Влияние поляризуемости лучше всего проявляется в относительной активности алкилйодидов и алкилфторидов. Несмотря на то что связь С-F несравненно сильнее поляризована, чем связь C-I, высоколежащие электроны атома йода более поляризуемы под действием нуклеофила. В результате алкилйодиды несопоставимо активнее алкилфторидов в реакциях алкилирования.
Большую роль играет также стабильность уходящей группы Х: чем она выше, тем легче протекает ее замещение (существует выражение «хорошая уходящая группа»). Например, спирты не реагируют с бромидом калия с образованием бромистых алкилов, поскольку ОН– – плохая уходящая группа. В присутствии же кислот, протонирующих спиртовый гидроксил, реакция протекает легко, поскольку в этом случае уходящей группой является нейтральная и устойчивая молекула воды:
Есть общее правило, которое легко запомнить: сильные основания (ОН–, Н– или СН3–) являются плохими уходящими группами, слабые основания – хорошими. Так, по легкости замещения некоторые из распространенных уходящих групп образуют следующий ряд:

Огромное влияние на легкость реакций нуклеофильного замещения оказывает нуклеофильность реагента. Последняя определяется как его способность отдавать электронную пару другим атомам. Нуклеофильность следует отличать от основности – способности отдавать электронную пару протону. Известно немало случаев, когда более слабое основание (например, CH3S– < CH3O–) является вместе с тем более сильным нуклеофилом (CH3S– > CH3O). Чаще всего такие отклонения характерны для нуклеофилов с большим ковалентным радиусом, т. е. хорошо поляризуемых. Другая причина отсутствия корреляции между ну-клеофильностью и основностью – стерические факторы. Например, диизопропиламид-анион [(CH3)2CH]2N– – более сильное основание, чем амид-ион NH2–. В то же время, в отличие от последнего, он практически полностью лишен нуклеофильных свойств (например, способности замещать галоген в связях С-Х или присоединяться к карбонильной группе) из-за сильного экранирования атома азота двумя изопропильными группами.
Нуклеофильность нельзя рассматривать отдельно от природы растворителя, в котором проводится реакция. В протонодонорных средах (вода, спирты, уксусная кислота и т. п.) анионы сильно сольватированы, что сильно уменьшает их нуклеофильность. При этом в большей степени сольватированы маленькие анионы с точечным или менее диффузным зарядом (H-, F, OH-). Например, анионы с большим радиусом легче сбрасывают сольватную оболочку и, в результате, характеризуются повышенной нуклеофильностью. Например, в ряду галогенид-ионов нуклеофильность уменьшается в ряду: I– > Br– > Cl– > F.
В апротонных диполярных растворителях (ацетон, диметилформамид, диметилсульфоксид и т. п.) картина меняется на обратную и более нуклеофильным оказывается анион меньшего размера и с большей плотностью заряда: F– > Cl– > Br– > I-.
Эти анионы в таких растворителях практически несольватированы, и в то же время они отделены от противоиона (обычно катиона щелочного металла), поскольку последний сам оказывается сольватированным в результате йон-дипольного взаимодействия с молекулой растворителя:
Реакциям нуклеофильного замещения часто благоприятствуют катализаторы, в качестве которых обычно применяются кислоты Льюиса. Все они так или иначе содействуют отщеплению уходящей группы. Выше уже говорилось о каталитической роли серной кислоты при замене спиртового гидроксила на галоген. Помимо протонов, такие кислоты Льюиса, как AlCl3, ZnCl2, BF3, HgCl2, катионы Ag+ и Сu+, координируясь с галогеном в алкилгалогенидах, способствуют его более легкому элиминированию:
Роль катализатора, как и растворителя, проявляется и в связи с более тонкими особенностями механизма реакции. Различают два основных механизма нуклеофильного замещения: мономолекулярный SN1 и бимолекулярный SN2. При мономолекулярном замещении процесс протекает в две стадии. На первой, более медленной, происходит диссоциация связи С-Х в молекуле субстрата, в результате чего образуются карбокатион и анион уходящей группы. На второй, быстрой стадии карбокатион соединяется с нуклеофилом с образованием конечного продукта замещения:
Поскольку на скорость-определяющей (лимитирующей) стадии в реакции занята только одна молекула субстрата, скорость всего процесса зависит лишь от концентрации субстрата и отвечает кинетическому уравнению первого порядка:
v = k [RX]
В отличие от этого при бимолекулярном замещении реакция протекает фактически в одну стадию: нуклеофил атакует углеродный атом субстрата с тыла, со стороны, противоположной той, где находится уходящая группа. Процесс образования новой связи и разрыв старой происходит синхронно, через рыхлое переходное состояние, в котором углеродный атом из состояния sp3 переходит в уплощенное состояние, близкое к sp2-гибридизации:
Скорость всего процесса, как легко заметить, в этом случае должна зависеть от концентрации как субстрата, так и реагента и отвечать уравнению реакции второго порядка:
Cуществует три основных доказательства реализации SN1 или SN2 механизма:
1) кинетическая зависимость скорости реакции от концентрации либо одного субстрата (SN1-механизм), либо от концентрации и субстрата, и нуклеофила (SN2-механизм);
2) обращение или сохранение стереохимической конфигурации углеродного атома, у которого протекает замещение. В случае SN2-реакции, как видно из приведенной выше схемы, нуклеофил занимает место, противоположное тому, где находилась уходящая группа, а остальные связи углерода как бы выворачиваются подобно зонтику при сильном ветре. Если углеродный атом, несущий уходящую группу, в молекуле субстрата хиральный, т. е. все четыре заместителя в нем разные, продукт реакции изменяет знак вращения на обратный. Это явление, открытое латвийским ученым Вальденом в начале ХХ в., получило название вальденовского обращения. В случае SN1-механизма вальденовского обращения в чистом виде не наблюдается, так как промежуточный плоский карбокатион с одинаковой вероятностью может атаковаться нуклеофилом и с одной и с другой стороны:
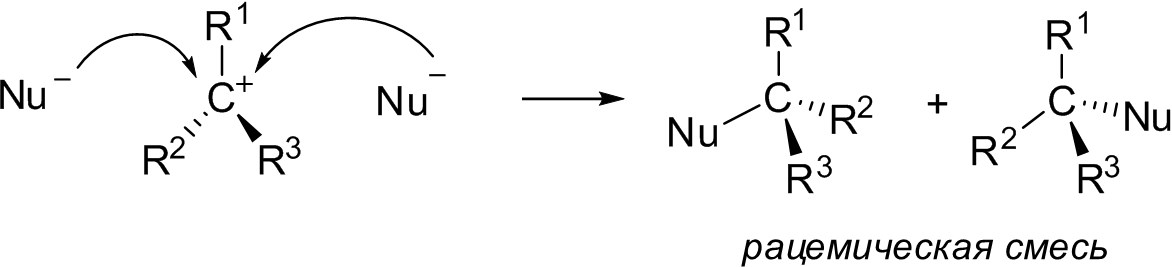
Впрочем, и при SN1-механизме возможно частичное обращение конфигурации. Это бывает в тех случаях, когда карбокатион образует с анионом тесную йонную пару, т. е. уходящая группа не уходит от карбокатионного центра слишком далеко, тем самым в некоторой степени мешая нуклеофилу подойти к углеродному атому со своей стороны. Кроме того, нередки случаи, когда реакция замещения протекает параллельно по обоим механизмам, и это также выражается в частичном обращении стереохимической конфигурации;
3) характерная зависимость реакционной способности субстрата от строения углеводородного радикала R. При реализации SN1-механизма, чем разветвленнее радикал R, тем легче протекает реакция. Для SN2-реакций картина прямо противоположная.
В случае SN2-реакций разветвленность алкильного радикала создает трудности для подхода нуклеофила. В случае же SN1-реакций такой проблемы нет, поскольку три заместителя при карбокатионном центре располагаются в одной плоскости и серьезных стерических препятствий для подхода нуклеофила не создают. Но главное же заключается в том, что третичные карбокатионы устойчивее вторичных, а последние в свою очередь устойчивее первичных – следствие делокализации положительного заряда за счет +I-эффекта алкильных групп:
Это обстоятельство существенно снижает активационный барьер для алкилгалогенидов изостроения и направляет для них реакцию по SN1-пути. Практически всегда реагируют по SN1-механизму метоксиметилхлорид CH3OCH2Cl и аллилгалогениды СН2=СН-СН2-Х, которые образуют резонансно-стабилизированные карбокатионы:
То же самое относится к трифенилметилхлориду (тритилхлориду), дифенилметилхлориду (бензгидрилхлориду) и бензилхлориду, реакционная способность которых изменяется в следующей последовательности (бензилхлорид частично реагирует и по SN2-механизму, который для него, однако, не является главным):
(C6H5)3C-Cl > (C6H5)2CH-Cl > C6H5CH2-Cl >> CH3-Cl
Реакции нуклеофильного замещения у насыщенного атома углерода сопровождаются рядом побочных процессов. Один из них – отщепление галогеноводорода от алкилгалогенида или воды от спиртов. При таком элиминировании продуктом реакции становится алкен. Различают два механизма элиминирования: E2 и Е1. Е2-Механизм реализуется как синхронный процесс, скорость которого зависит как от концентрации субстрата, так и нуклеофила:
Особенно легко подвергаются E2-реакциям вторичные и третичные алкилгалогениды. Так, изопропилбромид при действии этилата натрия в этаноле дает лишь 20 % продукта замещения, пропилен же образуется с выходом 80 %.

Третичные алкилгалогениды в этих условиях образуют практически полностью соответствующий алкен. Этому способствуют два обстоятельства. Во-первых, чисто статистически у трет-бутильного радикала в трет-бутилхлориде больше ?
Данный текст является ознакомительным фрагментом.