Глава 40 ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ МЕХАНИКИ
Глава 40
ПРИНЦИПЫ СТАТИСТИЧЕСКОЙ МЕХАНИКИ
§ 1. Экспоненциальная атмосфера
§ 2. Закон Больцмана
§ 3. Испарение жидкости
§ 4. Распределение молекул по скоростям
§ 5. Удельные теплоемкости газов
§ 6. Поражение классической физики
§ 1. Экспоненциальная атмосфера
Мы уже изучали некоторые свойства большого числа сталкивающихся атомов. Наука, которая занимается этим, называется кинетической теорией, и она описывает свойства вещества, рассматривая, как сталкиваются атомы. Мы утверждаем, что все свойства вещества в целом можно объяснить, рассматривая движение отдельных его частей.
Пока мы ограничимся случаем теплового равновесия, т. е. всего лишь подклассом всех явлений природы. Законы механики, применяемые в условиях теплового равновесия, получили название статистической механики, и в этой главе вы немного познакомитесь с некоторыми основными теоремами этой науки.
Одна теорема статистической механики вам уже известна. Согласно этой теореме, для любого движения при абсолютной температуре Т средняя кинетическая энергия каждого независимого движения (каждой степени свободы) равна 1/2kT. После этого нам становится кое-что известно о среднем квадрате скорости атомов. Теперь нам необходимо узнать чуть побольше о координатах атомов, чтобы выяснить, много ли их находится при тепловом равновесии в той или иной точке пространства, а также немного подробнее изучить распределение атомов по скоростям. Хотя мы знаем, чему равен средний квадрат скорости, мы все же не можем ответить на вопрос, сколько атомов обладают скоростью, в три раза большей, чем корень из среднего квадрата скорости, или скоростью, равной одной четверти корня из среднего квадрата скорости. А вдруг все атомы имеют одинаковую скорость?
Итак, вот два вопроса, на которые мы попытаемся дать ответ: 1) Как атомы располагаются в пространстве, когда на них действуют силы? 2) Каково распределение атомов по скоростям?
Заметим, что это два совершенно независимых вопроса и что распределение по скоростям всегда одинаково. Этого можно было ожидать после того, как мы выяснили, что средняя кинетическая энергия степени свободы всегда равна 1/2kT, независимо от того, какие силы действуют на молекулы. Распределение по скоростям молекул не зависит от сил, потому что силы не влияют на частоту столкновений.
Давайте начнем с примера распределения молекул в атмосфере, подобной той, в которой мы живем, но без ветра или других каких-либо возмущений.
Предположим, что мы имеем дело с довольно высоким столбом газа, находящегося в тепловом равновесии (не так, как в настоящей атмосфере; в ней, как известно, по мере подъема вверх становится холоднее). Укажем здесь, что нарушение равновесия в случае разницы температур на разных высотах можно продемонстрировать, поместив в столб газа металлический стержень так, что его концы соприкасаются с маленькими шариками (фиг. 40.1).
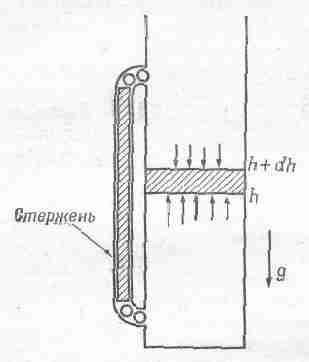
Фиг. 40. 1. Равновесие в атмосфере с постоянной температурой.
Давление на высоте h должно превосходить давление на высоте h+dh на вес заключенного между этими уровнями газа. Стержень и шарики выравнивают температуру.
Нижние шарики, получая от молекул газа энергию l/2kT, передают ее через стержень верхним шарикам и встряхивают их; верхние шарики в свою очередь будут встряхивать соприкасающиеся с ними наверху молекулы. В конце концов, конечно, температура на разных высотах гравитационного поля станет одинаковой.
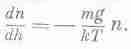
Нам предстоит найти закон, по которому происходит разрежение атмосферы по мере подъема вверх, когда температура на всех высотах одинакова. Если N — полное число молекул в объеме V газа с давлением Р, то PV=NkT, или Р=nkT, где n — число молекул в единичном объеме. Иначе говоря, если известно число молекул в единичном объеме, то известно и давление, и наоборот: давление и плотность пропорциональны друг другу, ведь температура в нашем случае постоянна. Но давление не может быть постоянным: с уменьшением высоты оно должно возрастать, потому что нижнему слою приходится, так сказать, выдерживать вес всех расположенных сверху атомов. Теперь можно определить, как давление меняется с высотой. Если на высоте h выделить площадку единичной площади, то на эту площадку снизу будет действовать сила, равная давлению Р. Если бы не было силы тяжести, то на площадку на высоте h+dh действовала бы сверху вниз точно такая же сила. Но в нашем случае это не так: действующая снизу сила должна превосходить силу, действующую сверху, на величину, равную весу газа, заключенного между слоями h и h+dh. На каждую молекулу действует сила тяжести mg, где g — ускорение силы тяжести. В интересующем нас слое находится ndh молекул. Это приводит к такому дифференциальному уравнению: Ph+dh-Ph=dP=-mgndh. Поскольку Р=nkT, а Т—постоянная, то можно избавиться или от Р, или от n. Исключим из уравнений Р; тогда получим
Это дифференциальное уравнение говорит нам, как убывает плотность по мере увеличения высоты.
Мы располагаем теперь дифференциальным уравнением для плотности частиц n, которая меняется с высотой, но меняется так, что производная плотности пропорциональна себе самой. Функция, производная которой пропорциональна себе самой,— это экспоненциальная функция и, значит, решение дифференциального уравнения имеет вид
n=n0e-mgh/kT. (40.1)
Здесь постоянная интегрирования n0— плотность на высоте h=0 (которую можно задать произвольно); с высотой плотность экспоненциально убывает.
Заметим, что если имеется несколько сортов молекул с разными массами, то число их убывает по разным экспонентам. Число более тяжелых молекул убывает с высотой быстрее, чем число легких молекул. Поэтому можно ожидать, что раз кислород тяжелее азота, то по мере подъема вверх относительное содержание азота в атмосфере (смеси азота и кислорода) будет возрастать. В нашей атмосфере, во всяком случае на доступных высотах, этого фактически не происходит, ибо вследствие воздушных возмущений газы вновь перемешиваются.
Ведь это же не изотермическая атмосфера. Тем не менее на больших высотах преобладают очень легкие газы, например водород, так как молекулы легких газов способны забраться на такую высоту, где все остальные экспоненты уже вымрут (фиг. 40.2).
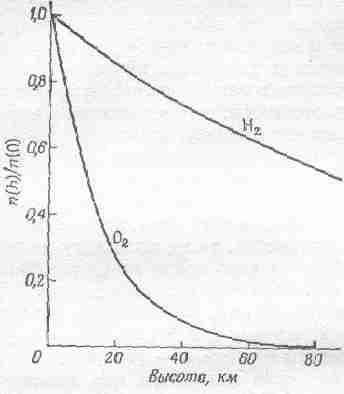
Фиг. 40.2. Нормированная плотность как функция высоты в гравитационном поле Земли для кислорода и водорода при постоянной температуре.
§ 2. Закон Болъцмаиа
Отметим здесь тот факт, что числитель показателя экспоненты в равенстве (40.1) — это потенциальная энергия, атома. Поэтому можно в нашем случае сформулировать закон следующим образом: плотность в каждой точке пропорциональна
e-п.э./kT
где п.э. — потенциальная энергия отдельного атома.
Возможно, что это случайность и этот закон справедлив только в частном случае однородного гравитационного поля. Однако можно показать, что это весьма общее утверждение. Предположим, что на молекулы газа действуют какие-то иные, не гравитационные, силы. Например, молекулы обладают электрическим зарядом, а тогда они реагируют на электрическое поле или на другой заряд, притягивающий их. А может быть, в результате взаимного притяжения атомов друг к другу или к стенкам, или к какому-нибудь твердому телу, или еще к чему-то существуют какие-то силы притяжения, которые зависят от взаимного расположения молекул и действуют на все молекулы. Предположим теперь для простоты, что все молекулы одинаковы и что сила действует на каждую отдельиую молекулу, так что полная сила, действующая на произвольно выделяемую часть газа, равна просто произведению числа молекул на силу, действующую на одну молекулу. Дело совсем упростится, если выбрать систему координат так, что сила F будет действовать вдоль оси х.
Так же, как и раньше, рассечем газ двумя параллельными плоскостями, промежуток между которыми равен dx. Тогда сила, действующая на каждый атом, умноженная на число атомов в 1 см3(обобщение прежнего nmg) и умноженная на dx, должна сбалансировать изменение давления: Fndx=dP=kTdn. Или, придав этому закону другую форму, которая пригодится позднее, запишем:
F=(kTd/dx)lnn (40.2)
Теперь заметим, что —Fdx — это работа, которую надо совершить для переноса молекулы из х в х+dx, и если сила F произошла из потенциала, т. е. работу можно описывать с помощью потенциальной энергии, то нужную нам величину можно считать изменением потенциальной энергии (п. э.). Отрицательное изменение потенциальной энергии — это произведенная работа Fdx, так что d(lnn)=-d(п. э.)/kT, или после интегрирования
n=(постоянная) е-п.э/kT. (40.3)
Таким образом, то, что нам удалось заметить в частном случае, справедливо вообще. (А что если F не происходит из потенциала? Тогда (40.2) просто-напросто не имеет решения. В этом случае, после того как какой-нибудь атом опишет замкнутый путь, вдоль которого полная работа не равна нулю, энергия либо прибавится, либо убавится и равновесие никогда не установится. Температурное равновесие невозможно, если внешние силы, действующие на газ, не консервативны.) Уравнение (40.3) известно под названием закона Болъцмана. Это еще один из принципов статистической механики: вероятность найти молекулу в заданной точке заданной пространственной конфигурации изменяется экспоненциально, причем показатель экспоненты состоит из потенциальной энергии в заданной пространственной конфигурации, взятой с обратным знаком и деленной на kT.
Таким образом, мы знаем кое-что о распределении молекул. Предположим, что в нашем распоряжении имеется плавающий в жидкости положительный ион; он притягивает окружающие его отрицательные ионы. Много ли их окажется на разных расстояниях от положительного иона? Если нам известно, как зависит от расстояния потенциальная энергия, то отношение чисел ионов на разных расстояниях определяется полученным нами законом. Этому закону можно найти еще много других применений.
§ 3. Испарение жидкости
В менее элементарной статистической механике пытаются решить следующую важную задачу. Предположим, что имеется совокупность притягивающихся друг к другу молекул и сила между любыми двумя молекулами, скажем i-й и j-й, зависит только от расстояния между ними rijи может быть представлена в виде производной от потенциальной энергии V(rij). На фиг. 40.3 показан возможный вид такой функции.
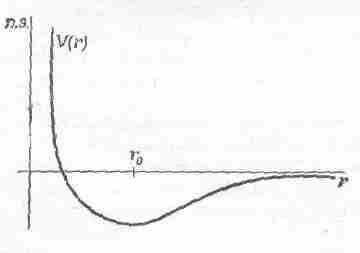
Фиг. 40. 3. Кривая потенциальной энергии для двух молекул. Потенциальная энергия зависит только от расстояний.
Если r>r0, то при сближении молекул энергия уменьшается, поэтому молекулы притягиваются; если же молекулы сближаются еще теснее, энергия очень резко возрастает, значит, на малых расстояниях молекулы сильно отталкиваются. Таково в общих чертах поведение молекул.

Предположим теперь, что мы заполнили этими молекулами какой-то ящик и хотим знать, как они там уместятся в среднем. На это даст ответ выражение ехр(-п. э./kT). В этом случае полная потенциальная энергия, если предположить, что молекулы взаимодействуют только попарно, равна сумме всех парных энергий (в более сложных случаях могут встретиться и тройные силы, но электрические силы, например, парные). Поэтому вероятность того, что молекулы образуют конфигурацию, характеризуемую заданными комбинациями расстояний rij, пропорциональна
Если температура очень высока, так что kT>>|V(r0)|, то экспонента почти всюду мала, и вероятность найти молекулу в том или ином месте почти не зависит от расстояния до других молекул, Рассмотрим случай двух молекул; в этом случае ехр (—п.э./kT) будет вероятностью найти молекулы на расстоянии rдруг от друга. Ясно, что вероятность максимальна тогда, когда потенциал наиболее отрицателен, а когда потенциал стремится к бесконечности, вероятность почти равна нулю (это происходит на очень малых расстояниях). Это означает, что у атомов газа нет шансов столкнуться друг с другом, уж очень сильно они отталкиваются. Но очень велики шансы найти эти молекулы (если отнести вероятность к единичному объему) вблизи точки r0. Здесь вероятность больше, чем в других точках, но насколько больше — это зависит от температуры. Если температура очень велика по сравнению с разностью энергий в точках r=r0и r=Ґ, то экспонента всегда почти равна единице. Это случай, когда средняя кинетическая энергия (она порядка kT) значительно превосходит потенциальную энергию. Силы тогда мало что значат. Но с падением температуры вероятность найти молекулы на расстоянии, близком к r0, резко возрастает по сравнению с вероятностью найти молекулы в любом другом месте; и в самом деле, если kT много меньше |V(r0)|, то около r0экспонента имеет довольно большой положительный показатель. Другими словами, при заданном объеме молекулы предпочитают быть на расстоянии минимальной энергии, а не очень далеко друг от друга. По мере падения температуры атомы сближаются, сбиваются в кучу, объединяются в жидкости, в твердые тела и молекулы, а если их подогреть, то они испаряются.
Если бывает необходимо точно описать, как происходит испарение, или вообще уточнить, как молекулы ведут себя в данных обстоятельствах, то поступать следует так. Прежде всего нужно как можно точнее узнать закон взаимодействия молекул V(r). Как это сделать — безразлично: можно вычислить потенциал с помощью квантовой механики или установить закон взаимодействия экспериментально. Но если даже закон взаимодействия молекул известен, нужно все же учесть, что дело идет о миллионах молекул и нам еще придется хватить горя при изучении функции ехр(—SVij/kT). Все же удивительно, что функция так проста и все как будто ясно, поскольку известен точный потенциал взаимодействия, а дело это оказывается невероятно сложным: трудность заключается в ужасающе большом числе переменных.
Но вопрос захватывающе интересен. Это один из примеров того, что называют «задачей многих тел», и он содержит много поистине увлекательных вещей. Одна-единственная формула, которую мы получим, решив задачу, должна содержать все детали, например переход газа в твердое состояние или возможные кристаллические строения твердого тела. Многие пытались ее сосчитать, но математические трудности уж очень велики, и дело не в трудности вывода общего закона, а просто в том, чтобы справиться с огромным числом переменных.
Вот и все, что касается распределения частиц в пространстве. На этом, собственно, и кончается классическая статистическая механика, ибо если нам известны силы, то в принципе мы можем найти пространственное распределение, а распределение скоростей находится сразу на все случаи жизни, оно не будет меняться от случая к случаю. Основная задача состоит в получении более конкретной информации из нашего формального решения: это и является основным занятием классической статистической механики.
§ 4. Распределение молекул по скоростям
Обсудим теперь распределение молекул по скоростям, потому что интересно, а иногда и полезно знать, какая часть молекул движется с той или иной скоростью. Чтобы выяснить это, можно использовать те знания, которые мы приобрели, когда изучали распределение газа в атмосфере. Мы считаем газ идеальным; мы предполагали это, пренебрегая взаимным притяжением атомов при расчете потенциальной энергии. В наш первый пример мы включили лишь потенциальную энергию силы тяжести. Если бы между атомами существовали взаимные силы, то нам, конечно, пришлось бы написать что-нибудь более сложное. Но мы по-прежнему будем предполагать, что между атомами никаких сил нет, и на момент даже забудем о столкновениях; потом мы попытаемся найти этому оправдание. Мы видим, что на высоте h находится гораздо меньше молекул, чем на высоте 0 (фиг. 40.4); согласно формуле (40.1), число их экспоненциально убывает с высотой.
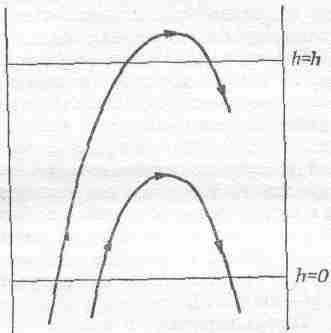
Фиг. 40.4. Высоты h достигают только те молекулы, скорость которых на высоте h=0 достаточно велика.
Но почему же на большей высоте меньше молекул? Разве не все молекулы, живущие на высоте 0, появляются на высоте h? Нет! Потому что на высоте 0 есть молекулы, движущиеся слишком медленно, и они не способны взобраться на потенциальную гору до высоты h. Вот и ключ к решению задачи о распределении молекул по скоростям; ведь, зная равенство (40.1), мы знаем число молекул, скорость которых слишком мала для достижения высоты h. Их ровно столько, чтобы создать нужное падение плотности при увеличении h.
Давайте сформулируем все поточнее: подсчитаем, сколько молекул проходит снизу вверх через плоскость h=0 (называя заданный уровень нулевой высотой, мы вовсе не считаем, что здесь пол, просто это удобнее нам для начала отсчета, и на отрицательной высоте может находиться газ). Эти молекулы газа движутся во всех направлениях, и некоторые из них проходят через нашу плоскость; таким образом, в любой момент сквозь плоскость снизу вверх проходит известное число молекул в секунду с заданной скоростью. Затем отметим следующее: если через uобозначить скорость, необходимую для того, чтобы подняться на высоту h (кинетическая энергия mu2/2=mgh), то число молекул в секунду, поднимающихся с нижней плоскости строго вверх и имеющих составляющую скорости, большую чем u, в точности равно числу молекул, пересекающих верхнюю плоскость с любой вертикальной составляющей скорости. Те молекулы, вертикальная скорость которых не превышает и, не
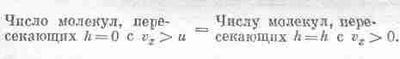
достигают верхней плоскости. Таким образом,
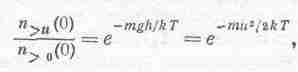
Но число молекул, пересекающих h с любой скоростью, большей нуля, меньше числа молекул, пересекающих нижний уровень с любой скоростью, большей нуля, хотя бы потому, что внизу больше атомов. Вот и все, что нам нужно. Мы уже знаем, что распределение молекул по скоростям на всех высотах одинаково, ведь мы уже выяснили, что температура во всей атмосфере одинакова. Но поскольку распределение скоростей всюду одинаково и число атомов, пересекающих нижний уровень, больше, то ясно, что отношение n>0(h) (числа атомов, пересекающих высоту h с положительной скоростью) и n>0(0) (числа атомов, пересекающих с положительной скоростью высоту 0) равно отношению плотностей на этих высотах, т. е. ехр(—mgh/kT). Но n>0(h)=h>u(0), поэтому
поскольку 1/2mu2=mgh. Теперь скажем это своими словами: число молекул, пересекающих за 1 сек единичную площадь
на высоте 0 с вертикальной составляющей скорости, превышающей и, равно произведению числа молекул, пересекающих эту площадку со скоростью, большей нуля, на ехр(-mu2/2kT).
Это верно не только для произвольной высоты 0, но и для любой другой высоты, поэтому распределение по скоростям одинаково повсюду! (Окончательный результат не включает высоты h, она появляется только в промежуточных рассуждениях.) Это общая теорема о распределении по скоростям. В ней утверждается, что если в столбе газа просверлить крохотную дырочку, ну совсем малюсенькую, так что столкновения там будут редки и длина пробега молекул между столкновениями будет много больше диаметра дырочки, то молекулы будут вылетать из нее с разными скоростями, но доля частиц, вылетающих со скоростью, превышающей и, равна ехр(-mu2/2kT).
Теперь вернемся к вопросу о том, можно ли пренебрегать столкновениями. Почему это не имеет значения? Мы могли бы повторить все наши доводы, используя не конечную высоту h, а бесконечно малую высоту h, столь малую, что для столкновений между высотами 0 и h было бы слишком мало места. Но это не обязательно: наши доводы, очевидно, основаны лишь на анализе значений энергий и на сохранении энергии; при столкновениях же происходит обмен энергиями среди молекул. Но нам довольно безразлично, следим ли мы за одной и той же молекулой, раз происходит лишь обмен энергиями с другой молекулой. И получается, что если мы даже сделаем это достаточно тщательно (а такую работу тщательно проделать, конечно, труднее), то результат будет тот же.
Интересно, что найденное нами распределение по скоростям имеет вид
n>u~e-к.э./kT. (40.4)
Этот способ описания распределения по скоростям —когда подсчитывается число молекул, проходящих через выделенную площадку с заданной минимальной z-составляющей скорости,— отнюдь не самый удобный. Например, чаще хотят знать, сколько молекул в заданном объеме газа движется, имея z-составляющую скорости между двумя заданными значениями, а это, конечно, из (40.4) сразу не получишь. Поэтому придадим нашей формуле удобную форму, хотя то, что мы получили, — это весьма общий результат. Заметим, что невозможно утверждать, что любая молекула в точности обладает той или иной наперед заданной скоростью; ни одна из них не движется со скоростью, в точности равной 1,7962899173 м/сек. Итак, чтобы придать нашему утверждению какой-то смысл, мы должны спросить, сколько молекул можно найти в заданном интервале скоростей. Нам придется говорить о том, как часто встречаются скорости в интервале между 1,796 и 1,797 и т. п. Выражаясь математически, пусть f(u)du будет долей всех молекул, чьи скорости заключены в промежутке u и u+du, или, что то же самое (если du бесконечно мало), долей всех молекул, имеющих скорость и с точностью до du. На фиг. 40.5 представлена возможная форма функции f(u), а заштрихованная часть ширины du и средней высоты f(u) — это доля молекул f(u)du. Таким образом, отношение площади заштрихованного участка ко всей площади под кривой равно относительному числу молекул со скоростью и внутри отрезка du.
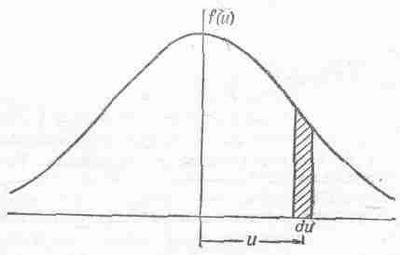
Фиг. 40.5. Функция, распределения скоростей.
Заштрихованная площадь равна f(u)du — это относительное число частиц, скорости которых заключены внутри отрезка du около точки u.
Если определить f(u) так, что относительное число молекул будет просто равно площади заштрихованного участка, то полная площадь под кривой — это все 100% молекул, т. е.

Теперь остается только найти это распределение, сравнив его с результатом доказанной ранее теоремы. Сначала надо выяснить, как выразить через f(u) число молекул, проходящих за 1 сек через заданную площадку со скоростью, превышающей u?
Это число не равно интегралу
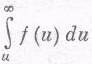 (хотя это первое, что приходит в голову), ведь нас интересует число молекул, проходящих через площадку за секунду. Более быстрые молекулы будут пересекать площадку, так сказать, чаще, чем более медленные, поэтому, чтобы найти число проходящих молекул, надо умножить плотность молекул на скорость. (Мы уже обсуждали это в предыдущей главе, когда подсчитывали число столкновений.)
(хотя это первое, что приходит в голову), ведь нас интересует число молекул, проходящих через площадку за секунду. Более быстрые молекулы будут пересекать площадку, так сказать, чаще, чем более медленные, поэтому, чтобы найти число проходящих молекул, надо умножить плотность молекул на скорость. (Мы уже обсуждали это в предыдущей главе, когда подсчитывали число столкновений.)
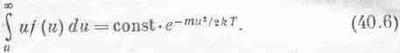
Полное число молекул, проходящих через поверхность за время t, равно числу молекул, способных достигнуть поверхности, а это молекулы, проходящие к поверхности с расстояния ut. Таким образом, число молекул, достигающих площадки, определяется не просто числом молекул, движущихся с данной скоростью, а равно этому числу, отнесенному к единице объема, и умноженному на расстояние, которое они пройдут, прежде чем достигнут площадки, сквозь которую они, по-видимому, должны пройти, а это расстояние пропорционально и. Значит, нам предстоит вычислить интеграл от произведения и на f(u)du, взятый от и до бесконечности, причем мы уже знаем, что этот интеграл обязательно должен быть пропорционален ехр(-mu2/2kT), а постоянную пропорциональности еще надо определить:
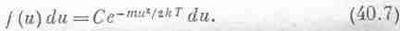
Если теперь продифференцировать интеграл по и, то мы получим подынтегральное выражение (со знаком минус, потому что и — это нижний предел интегрирования), а дифференцируя правую часть равенства, мы получим произведение и на экспоненту (и на некоторую постоянную). Сократим в обеих частях и, и тогда
Мы оставили в обеих частях равенства du, чтобы помнить, что это распределение; оно говорит нам об относительном числе молекул, имеющих скорость между u и u+du.
Постоянная С должна определиться из условия равенства интеграла единице в согласии с уравнением (40.5). Можно доказать, что
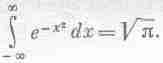
Используя это обстоятельство, легко найти С=Ц(m/2pkT).
Поскольку скорость и импульс пропорциональны, можно утверждать, что распределение молекул по импульсам, отнесенное к единице импульсной шкалы, также пропорционально ехр(-к.э./kT). Оказывается, что эта теорема верна также в теории относительности, если только формулировать ее в терминах импульсов, тогда как в терминах скоростей это уже не так; поэтому сформулируем все в терминах импульсов:
f(p)dp=ce-к.э./kTdp. (40.8)
Это значит, что мы установили, что вероятности, определяемые энергиями разного происхождения (и кинетической и потенциальной), в обоих случаях выражаются одинаково: ехр(-энергия/kT); таким образом, наша замечательная теорема приобрела форму, весьма удобную для запоминания.
Однако пока мы говорили только о «вертикальном» распределении скоростей. Но мы можем спросить, какова вероятность того, что молекула движется в другую сторону? Конечно, эти распределения связаны друг с другом и можно получить полное распределение, исходя из какого-то одного, ведь полное распределение зависит только от квадрата величины скорости, а не от ее z-составляющей. Распределение по скоростям не должно зависеть от направления и определяться только функцией u2 — вероятностью величины скорости. Нам известно распределение z-составляющей, и мы хотим получить отсюда распределение других составляющих. В результате полное распределение по-прежнему пропорционально ехр(-к.э./kT), только теперь кинетическая энергия состоит из трех частей: mv2x/2, mv2y/2 и mv2z/2, суммируемых в показателе экспоненты. А можно записать это и в виде произведения:
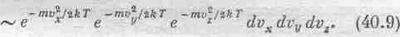
f(vx,, vy, vz) dvx dvy dvz~
Вы можете убедиться в том, что эта формула верна, ибо, во-первых, распределение зависит только от v2 и, во-вторых, вероятности данных vгполучаются после интегрирования по всем vxи vyи это должно привести к (40.7). Но обоим этим требованиям удовлетворяет только функция (40.9).
§ 5. Удельные теплоемкости газов
Посмотрим теперь, как можно проверить теорию и оценить, насколько хороша классическая теория газов. Мы уже говорили, что если U—внутренняя энергия N молекул, то формула pV=NkT=(g-1)U иногда и для некоторых газов может оказаться правильной. Мы знаем, что для одноатомного газа правая часть равна 2/3 кинетической энергии движения центров масс атомов. В случае одноатомного газа кинетическая энергия равна внутренней энергии, поэтому g-1 == 2/з.
Но предположим, что мы столкнулись с более сложной молекулой, которая может вращаться и колебаться, и предположим (в классической механике это так), что энергии внутренних движений также пропорциональны kT. Поэтому при заданной температуре молекула, кроме кинетической энергии kT, имеет внутреннюю энергию колебания и вращения. Тогда полная энергия U включает не только кинетическую энергию, но и вращательную энергию и мы получаем другие значения у. Наилучший способ измерения g — это измерение удельной теплоемкости, характеризующей изменение энергии при изменении температуры. К этому способу мы еще вернемся, а пока предположим, что нам удалось экспериментально определить g с помощью кривой PVg , соответствующей адиабатическому сжатию.
Попробуем вычислить g для ряда частных случаев. Прежде всего для одноатомных газов полная энергия U есть не что иное, как кинетическая энергия, и в этом случае, как мы уже знаем, g равно 5/3. В качестве примера двухатомных газов рассмотрим кислород, водород, пары иода и т. д. и предположим, что двухатомный газ можно представить как собрание пар атомов, между которыми действуют силы, похожие на те, что изображены на фиг. 40.3. Можно также предположить, и оказывается, что это вполне законно, что при температурах; обычных для диатомных газов, пары атомов стремятся удалиться друг от друга на расстояние r0 (расстояние минимума потенциальной энергии). Если бы это было не так, и вероятность не очень сильно зависела от удаления от равновесной конфигурации, то мы обнаружили бы, что кислород есть смесь сравнимых количеств O2 и одиночных атомов кислорода. А мы знаем, что в кислороде присутствует очень мало одиночных атомов кислорода, а это означает, что глубина потенциальной ямы значительно больше kT, и это как раз мы и предполагали. Но раз атомы, составляющие молекулу, прочно закреплены на расстоянии r0, то нам понадобится лишь часть потенциальной кривой вблизи минимума, которую в этом случае можно приближенно заменить параболой. Параболический потенциал соответствует гармоническому осциллятору, и, в самом деле, отличной моделью молекулы кислорода могут служить два соединенных пружинкой атома.
Но чему же равна полная энергия молекулы при температуре Т? Мы знаем, что кинетическая энергия каждого из атомов равна 3/2 kT, так что кинетическая энергия обоих атомов равна 3/2kT +3/2kT. Можно распределить эту энергию иначе: тогда те же самые 3/2 плюс 3/2 будут выглядеть как кинетическая энергия центра масс (3/2), кинетическая энергия вращения (2/2) и кинетическая энергия колебаний (1/2). Известно, что на долю кинетической энергии колебаний приходится 1/2, потому что это одномерное движение, а каждой степени свободы соответствует l/2kT. Обращаясь к вращениям, мы можем выделить две оси вращения, что соответствует двум независимым движениям. Мы представляем себе атомы в виде точек, которые не могут вращаться вокруг соединяющей их линии. Но на всякий случай запомним о таком предположении, потому что если мы упремся где-то в тупик, то, может быть, здесь обнаружится корень зла. Нас должен интересовать еще и другой вопрос: чему равна потенциальная энергия колебаний, велика ли она? Средняя потенциальная энергия гармонического осциллятора равна средней кинетической энергии, т.е. также l/2kT. Полная энергия молекулы U = 7/2kT, или kT=2/7U на атом. Это означает, что g равно 9/7, а не 5/3, т. е. g=1,286. Можно сравнить эти числа с действительно измеренными значениями g, приведенными в табл. 40.1. Взгляните сначала на гелий; это одноатомный газ, и значение g очень близко к 5/3; отклонение от этого значения, вероятно, есть просто следствие экспериментальных неточностей, хотя при столь низких температурах между атомами могут появиться силы взаимодействия. Криптон и аргон — еще два одноатомных газа — также дают согласующиеся значения в пределах ошибки эксперимента.
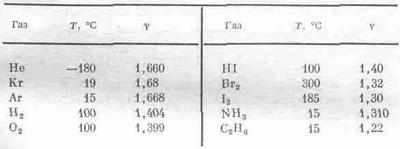
Таблица 40.1 · ИЗМЕРЕННЫЕ ЗНАЧЕНИЯ g ДЛЯ РАЗЛИЧНЫХ ГАЗОВ
Перейдем к двухатомным газам. Тут же обнаружится, что значение gдля водорода, равное 1,404, не согласуется с теоретическим значением 1,286. Очень близкое значение дает и кислород, 1,399, но с теоретическим это снова не согласуется. Для йодистого водорода g равно просто 1,40. Начинает казаться, что мы нашли общий закон: для двухатомных молекул gравно 1,40. Но нет, поглядите дальше. Для брома мы получаем 1,32, а для иода 1,30. Поскольку 1,30 довольно близко к 1,286, то можно считать, что экспериментальное значение gдля иода согласуется с теоретическим, а кислород представляет собой исключение. Это уже неприятно. То, что верно для одной молекулы, неверно для другой, и нам, по-видимому, надо проявить хитроумие, чтобы объяснить это.
Давайте рассмотрим еще более сложные молекулы, состоящие из большого числа частей, например С2Н6 — этан. Молекула атома состоит из восьми разных атомов, и все они качаются и вращаются в самых разных комбинациях, так что полная величина внутренней энергии должна складываться из огромного числа kT, по крайней мере 12kT только для одной кинетической энергии, поэтому g-1 должно быть очень близко к нулю, a g почти в точности равно единице. И действительно, значение g для этана меньше, чем в предыдущих случаях, но 1,22— не так
уж мало, во всяком случае, больше l1/12, чему должно быть
равно g, если учесть только кинетическую энергию. Этого вообще нельзя понять!
Ну а дальше совсем плохо, ибо двухатомную молекулу нельзя рассматривать как абсолютно жесткую, даже в пределе. Даже если связь между атомами так сильна, что они не могут и пошевелиться, все равно нужно считать, что они колеблются. Колебательная энергия всегда равна kT, поскольку она не зависит от силы связи. Но если представить себе двухатомную молекулу абсолютно жесткой, остановить колебания и выбросить эту степень свободы, то мы получим U=5/2 kT и g=1,40 для двухатомных газов. Казалось бы, это подходит и для Н2, и для O2. Но вопрос по-прежнему остается открытым, потому что g и для кислорода, и для водорода зависит от температуры! На фиг. 40.6 показаны результаты нескольких измерений. Для Н2 значение g изменяется от 1,6 при
-185°С до 1,3 при 2000°С. В случае водорода изменения g еще больше, но и в случае кислорода g явно стремится возрасти при падении температуры.
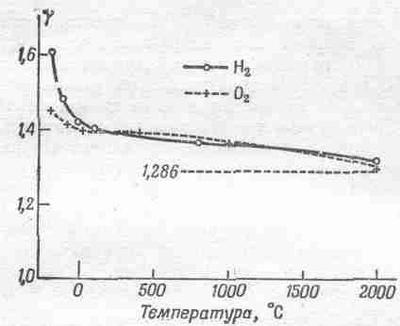
Фиг. 40.6. Экспериментальные значения g как функция температуры для водорода и кислорода.
Классическая теория предсказывает не зависящее от температуры значение g=1,286.
§ 6. Поражение классической физики
Итак, приходится сказать, что мы натолкнулись на трудности. Можно соединить атомы не пружинкой, а чем-нибудь другим, но оказывается, что это только увеличит значение g. Если пустить в ход другие виды энергии, то вопреки фактам g очень приблизится к единице. Все известное нам из классической теоретической физики только ухудшает положение. Нам известно, например, что каждый атом содержит электроны, и атомные спектры обязаны своим существованием внутренним движениям электронов; каждый электрон должен иметь по крайней мере l/2kT кинетической энергии и еще кое-что из потенциальной, а когда все это складывается, то g становится еще меньше. Просто смешно. И явно что-то не так.
Первая замечательная работа по динамической теории газов была сделана Максвеллом в 1859 г. Исходя из идей, с которыми мы только что познакомились, он сумел точно объяснить очень много известных явлений, таких, как закон Бойля, теорию диффузии, вязкость газов и другие вещи, о которых мы еще будем говорить в следующей главе. Подводя итог всем этим великим достижениям, он писал: «Наконец, установив необходимое соотношение между поступательным и вращательным движением несферических частиц (он имел в виду теорему о l/2kT), мы доказали, что в системе из таких частиц не может выполняться известное соотношение между двумя теплоемкостями». Он говорит здесь о g (позднее мы увидим, что эта величина связана с двумя разными способами измерения удельной теплоемкости) и замечает, что никто не в состоянии дать верного ответа.
В прочитанной десять лет спустя лекции он сказал: «Я должен изложить Вам то, что я считаю наибольшей трудностью, стоящей перед молекулярной теорией». Это было первое указание на ложность законов классической физики, первое предчувствие того, что существует нечто, необъясненное с самого начала, ибо опыту противоречила строго доказанная теорема.
Примерно в 1890 г. Джинс заговорил вновь об этой загадке. Часто приходится слышать, что физики конца девятнадцатого столетия были уверены в том, что им известны все существенные законы природы и дело стоит лишь за тем, чтобы получить нужные числа с максимальным числом десятичных знаков. Кто-то это сказал, а остальные повторяют. Но если покопаться в физических журналах тех лет, то станет ясно, что почти каждый из них в чем-нибудь да сомневался. Джинс говорил об этой проблеме как о загадочном явлении, из которого как будто бы следует, что по мере падения температуры некоторые виды движения «замерзают».
Если бы мы могли предположить, что колебаний при низких температурах нет и возникают они только при высоких температурах, то можно было бы представить существование такого газа, у которого при очень низкой температуре колебательного движения нет совсем, так что g=1,40, а при высоких температурах возникают колебания и, следовательно, g убывает. То же самое можно предположить и о вращениях. Если бы можно было избавиться от вращений, скажем, «заморозить» их, понизив достаточно температуру, то стало бы понятно, почему при низких температурах для водорода g приближается к 1,66. Но как же понять все это? Конечно, оставаясь в рамках классической механики, «замерзающих» движений нельзя объяснить. Все стало на свои места лишь после открытия квантовой механики.
Мы сформулируем без доказательства основные результаты статистической механики, построенной на основе квантовой механики. Напомним, что, согласно квантовой механике, связанная потенциалом система, например осциллятор, имеет дискретный набор уровней энергии, т. е. состояний с различной энергией. Возникает вопрос: как модифицировать статистическую механику, чтобы привести ее в согласие с квантовой механикой? Обратите внимание на интересную деталь: хотя большинство задач квантовой механики сложнее соответствующих задач классической физики, проблемы статистической механики решаются с помощью квантовой теории много проще!
Простенький результат классической механики, что n= n0ехр(-энергия/kT), становится в квантовой теории весьма важной теоремой: если набор молекулярных состояний характеризуется энергиями Е0, Е1, e2, ..., Еi, ..., то в случае теплового равновесия вероятность найти молекулу в состоянии с энергией Еiпропорциональна ехр(-Ei/kT). Так определяется вероятность пребывания в различных состояниях. Иначе говоря, относительный шанс — вероятность нахождения в состоянии Е1по сравнению с вероятностью нахождения в состоянии Е0равен


это, конечно, то же самое, что и
потому что Р1=n1/N, а Р0=n0/N. Таким образом, состояния с большей энергией менее вероятны, чем состояние с меньшей энергией. Отношение числа атомов в верхнем состоянии к числу атомов в нижнем состоянии равно е в степени (разность энергий, деленная на kT,с обратным знаком) — очень простая теорема.
Обратим внимание на то, что уровни энергии гармонического осциллятора отстоят друг от друга на равных расстояниях. Припишем низшему уровню энергию Е0=0 (на самом деле эта энергия немного отличается от нуля, но сдвиг всех уровней на одну и ту же величину не имеет значения), тогда энергия следующего уровня E1=hw, затем следует 2hw, 3hw) и т. д.
А теперь посмотрим, что из этого получится. Предположим, что мы изучаем колебания двухатомной молекулы, которую можно сейчас считать гармоническим осциллятором. Каковы относительные шансы найти молекулу в состоянии Е1, а не в состоянии Е0? Ответ: Отношение шанса найти молекулу в состоянии Е1 к шансу найти эту молекулу в состоянии Е0равно ехр(-hw/kT}. Предположим, что kT много меньше hw, т. е. мы находимся в области низких температур. Тогда вероятность обнаружить состояние e1чрезвычайно мала. Практически все молекулы находятся в состоянии Е0. Если изменить температуру, но по-прежнему поддерживать ее очень малой, то шанс найти молекулу в состоянии Е1=hwпо-прежнему бесконечно мал — энергия осциллятора все еще почти равна нулю; она не изменяется с температурой, пока температура остается много меньше hw. Все осцилляторы находятся в низшем состоянии, их движение эффективно «заморожено», и они не дают вклада в теплоемкость. С помощью данных табл. 40.1 можно установить, что при 100°С, а это равно 373?К (абсолютной температуры), kT много меньше колебательной энергии молекул кислорода и водорода, но сравнимо с колебательной энергией иода. Причина такой разницы в том, что атомы иода гораздо тяжелее атомов водорода и, хотя силы, действующие менаду атомами иода и водорода, сравнимы, молекула иода столь тяжела, что собственная частота ее колебаний чрезвычайно мала по сравнению с собственной частотой водорода. При комнатной температуре kT таково, что hwводорода больше kT, а hw иода — меньше. Поэтому классическую колебательную энергию можно обнаружить только у иода.
Если увеличивать температуру газа, начав с очень малых значений Т, когда почти все молекулы находятся в их низшем состоянии, то появляется ощутимая вероятность найти молекулу во втором состоянии, затем в следующем за ним и т. д. Когда много состояний получают заметную вероятность, газ ведет себя более или менее так, как того требует классическая физика, ведь в этом случае систему квантовых состояний трудно отличить от непрерывного распределения энергии, и система может обладать почти любой энергией. Таким образом, при повышении температуры мы снова попадаем в область классической физики, как это видно из фиг. 40.6. Аналогично можно показать, что точно так же квантуются и вращательные состояния атомов, но эти состояния размещены так тесно, что обычно kT больше расстояния между уровнями. В этом случае возбуждено сразу много уровней и вращательная кинетическая энергия системы ведет себя классически. Лишь водород при комнатных температурах ведет себя иначе.
Это первый случай, когда из сравнения с экспериментом обнаружилось, что с классической физикой что-то неблагополучно, мы искали способы уладить все трудности в квантовой механике тем самым путем, каким это происходило на самом деле. Прошло примерно лет 30 или 40, пока не была обнаружена еще одна трудность, и снова в статистической механике, но на этот раз в механике фотонного газа. Новая задача была решена Планком в первые годы нашего столетия.

* Чтобы вычислить этот интеграл, положим
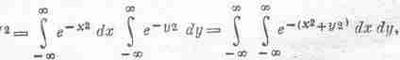
Тогда
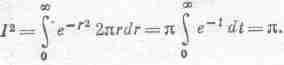
а это двойной интеграл в xy-плоскости. Но его можно вычислить и в полярных координатах:
Более 800 000 книг и аудиокниг! 📚
Получи 2 месяца Литрес Подписки в подарок и наслаждайся неограниченным чтением
ПОЛУЧИТЬ ПОДАРОКЧитайте также
1. Общие идеи и основные принципы
1. Общие идеи и основные принципы Понятие вероятности играло важную роль в первых физических трактовках волновой механики. Чувствовалось, что возникла общая теория, в которой все законы новой механики имеют вероятностный характер. К этой теории, внешне очень новой и
Физические принципы против логических структур
Физические принципы против логических структур Много лет я убеждался в том, что математика и физика подчиняются определенной диалектике взаимоотношений. Физика — не просто бессмысленная, произвольная последовательность диаграмм Фейнмана и симметрий, а математика —
9. Основы механики
9. Основы механики Механикой называют раздел физики, в котором изучается механическое движение материальных тел. Под механическим движением понимают изменение положения тела или его частей в пространстве с течением времени.Для медиков этот раздел представляет интерес
ОБЩИЕ ПРИНЦИПЫ
ОБЩИЕ ПРИНЦИПЫ В определенном смысле детекторы — и ATLAS, и CMS — представляют собой логическое развитие того пути, которое Галилей и другие ученые начали несколько столетий назад. Тогда с изобретения микроскопа началось развитие техники, которая позволяла физикам
5. Принципы неопределенности
5. Принципы неопределенности Мои попытки связаться со Стивеном после Испании были безрезультатны. Его мать сказала, что он уже вернулся в Кембридж и что дела с его здоровьем очень плохи. Приближалась осень, и я готовилась к отъезду из дома; меня ожидал новый жизненный этап
Глава 4 От механики Ньютона до электродинамики Максвелла
Глава 4 От механики Ньютона до электродинамики Максвелла Мне не стоило большого труда отыскание того, с чего следует начинать, так как я уже знал, что начинать надо с самого простого и доступного пониманию… Рене Декарт «Рассуждении о методе» Сейчас нам придется
Принципы построения
Принципы построения Попытки решить возникшие проблемы чрезвычайно активными были в начале XX века. В результате в 1905 году была окончательно сформулирована специальная теория относительности (далее будем обозначать ее СТО) и представлена одновременно в работах Альберта
Принципы построения ОТО
Принципы построения ОТО Не природа …сообразуется с принципами, а наоборот, принципы верны лишь постольку, поскольку они соответствуют природе… Фридрих Энгельс «Диалектика природы» Пришло время начать рассказ собственно об общей теории относительности, о принципах ее
Принципы Померанчука
Принципы Померанчука Впервые я встретился с И. Я. Померанчуком зимой 1947/48. Я учился в Университете на кафедре строения вещества на четвёртом курсе. Надо было подыскивать себе руководителя дипломной работы. Я хотел выбрать его из школы Ландау. (Такое допускалось на кафедре
ПРИНЦИПЫ РАЗДЕЛЕНИЯ ПОСРЕДСТВОМ ДИФФУЗИИ
ПРИНЦИПЫ РАЗДЕЛЕНИЯ ПОСРЕДСТВОМ ДИФФУЗИИ ОДНОСТУПЕНЧАТАЯ ДИФФУЗИЯ10.5. Как было указано в прошлой главе, скорость диффузии газа через идеальную пористую перегородку обратно пропорциональна квадратному корню из его молекулярного веса. Так, если газ, состоящий из двух
Принципы научного администрирования
Принципы научного администрирования Майкл Б. ШИМКИН Почти в каждой биологической лаборатории на стене висит портрет Луи Пастера с двумя кроликами в руках (вид у кроликов довольно жалкий). Тридцать лет назад ни один ученый, достаточно знаменитый, чтобы быть изображенным
Глава первая Немного механики
Глава первая Немного механики Скала Эдисона Незадолго до смерти знаменитый американский изобретатель Эдисон пожелал отличить самого сметливого юношу своей страны, назначив ему щедрую денежную поддержку для дальнейшего образования. Со всех концов республики были